روش پارامتردار کردن ۳ (PM3) تقریبأ همان معادلات به کار رفته در روش AM1 را فقط با یک مجموعهی بهینه در محاسبهی مولکولها به کار میبرد. روش PM3 برای محاسبهی ترکیبات رایج است. این روش در تخمین زوایای پیوند هیدروژنی خیلی دقیقتر از روش AM1 میباشد، در حالی که برای محاسبهی انرژی پیوندهای هیدروژنی، روش AM1 اطلاعات دقیقتری را به ما میدهد. روشهای PM3 و AM1 بیشتر از سایر روشهای نیمه تجربی رایج میباشد و دلیل آن به وجود الگوریتمهایی برای دخالت دادن اثرات حلال در این محاسبات برمیگردد.
برای روش PM3 نیز مزایا و معایبی را میتوان اشاره کرد. تمامی گرماهای تشکیل محاسبه شده با این روش دقیقتر از مقادیر محاسبه شده با روش AM1 میباشد. این روش، مولکولهای با ظرفیت زیاد را بهتر مدلسازی میکند. پیشبینی انرژی سد چرخشی حول پیوند C-N در پپتیدها با این روش نتایج ضعیفی را به ما میدهد. پیوندهای بین سیلیسیم و اتمهای هالید در این روش بسیار کوچک به دست میآید. همچنین روش AM1 پیشبینی ناصحیحی از حالتهای الکترونیکی ترکیبات ژرمانیوم دارد. این روش در محاسبات مولکولی، همیشه اتم نیتروژن sp3 را به صورت نیتروژن هرمی در نظر میگیرد. محاسبات تمایل به جذب پروتون با این روش به خوبی صورت نمیپذیرد. تعدادی از ترکیبات چند حلقهای بعد از شبیهسازی با این روش معمولأ به صورت مسطح دیده نمیشود. بار پیشبینی شده بر روی اتم نیتروژن با این روش مقدار صحیحی نیست. فواصل غیر پیوندی، خیلی کوتاهتر از مقدار واقعی به دست میآید. پیوندهای هیدروژنی محاسبه شده خیلی کوتاه هستند، در حالی که جهتگیری آنها معمولأ درست میباشد. در کل، این روش پیشبینی انرژیها و طول پیوندها را خیلی دقیقتر از روش AM1 انجام میدهد.
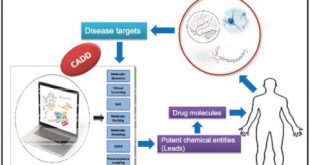
روشهای نیمه تجربی نتایج به اندازهی کافی دقیقی را برای استفاده به دست میدهد، مخصوصأ برای مولکولهای آلی با محاسبات نیازمند به PC با مشخصات بالا، این روشهای نیمه تجربی بسیار خوب عمل میکنند. این روشها به طور کلی برای پیشبینی ساختارهای هندسی مولکولها و انرژیها مناسب میباشند. روشهای نیمه تجربی توانایی پیشبینی مدهای ارتعاشی و ساختارهای حد واسط را دارند، اما نتایج حاصل از آنها نسبت به روشهای ab initio دقیق نمیباشند. محاسبات نیمه تجربی به طور کلی اطلاعات ضعیفی را در ارتباط با نیروهای بین مولکولی و واندروالس به دلیل فقدان مجموعه های پایهی نفوذی به دست میدهد.